Mit Ihrer Hilfe fördern wir Forschung
– damit medizinischer Fortschritt alle erreicht.

Spenden & aktiv werden
– damit medizinischer Fortschritt alle erreicht.

Gemeinsam Zukunft schaffen
Seltene Erkrankungen betreffen vor allem die Schwächsten in unserer Gesellschaft. Jedes Jahr sterben allein in Deutschland mindestens 1.500 Kinder an einer Seltenen Erkrankung. Selten ist dabei nur das jeweilige Krankheitsbild: Mindestens 4 Millionen Menschen sind hierzulande von einer Seltenen Erkrankung betroffen. Mangels Forschung fehlen wirksame Behandlungsansätze und Medikamente.
Das will die Eva Luise und Horst Köhler Stiftung durch gezielte Forschungsförderung, Netzwerk- und Öffentlichkeitsarbeit ändern. Wir fördern Forschung zu Seltenen Erkrankungen und investieren gezielt in die dafür notwendigen Strukturen. Als Initiatorin der Forschungsinitiative Alliance4Rare gestalten wir gemeinsam mit Partnerinnen und Partnern aus Forschung und Zivilgesellschaft ein Zukunftsmodell für die pädiatrische Forschung in Deutschland. Mit Ihrer Hilfe lassen wir die ‚Medizin von morgen‘ Wirklichkeit werden und sorgen dafür, dass der medizinische Fortschritt alle erreicht!
Wussten Sie, dass...

Die Stiftung
Wer wir sind
Seit 2006 engagieren sich die Menschen hinter der Eva Luise und Horst Köhler Stiftung aus tiefer Überzeugung für eine bessere medizinische Versorgung von Kindern, Jugendlichen und Erwachsenen mit Seltenen Erkrankungen.
Forschung

Sie sehen gerade einen Platzhalterinhalt von Youtube. Um auf den eigentlichen Inhalt zuzugreifen, klicken Sie auf die Schaltfläche unten. Bitte beachten Sie, dass dabei Daten an Drittanbieter weitergegeben werden.
Mehr InformationenAktuelles

Ausschreibungen
Ausschreibung der Alliance4Rare: Förderung von Forschungsprojekten zu Seltenen Erkrankungen
Die Alliance4Rare - Forschungsinitiative für Kinder mit Seltenen Erkrankungen schreibt erneut bis zu drei Forschungsprojekte mit einer Laufzeit von maximal 3 Jahren und einem jeweiligen Förderrahmen von maximal 150.000 Euro/…
Forschung
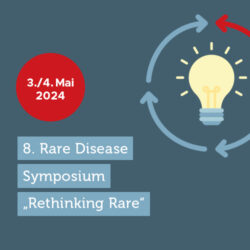
8. Rare Disease Symposium: „Rethinking Rare“
"Rethinking Rare“ – dazu laden wir im Rahmen des 8. Rare Disease Symposiums am 3./4. Mai 2024 in Berlin sehr herzlich ein. Bringen Sie sich ein beim Nach-, Weiter- und…

Forschung
Verleihung des 16. Eva Luise Köhler Forschungspreises für Seltene Erkrankungen
Eva Luise Köhler verleiht am Freitag, 3. Mai 2024 ab 18.00 Uhr in der Berlin-Brandenburgischen Akademie der Wissenschaften den nach ihr benannten Forschungspreis für Seltene Erkrankungen. Die Anmeldung ist ab…

Seltene Erkrankungen
Warum wir helfen
Selten sind viele. Unser Einsatz schenkt Betroffenen Hoffnung auf eine gesündere Zukunft und kommt letztendlich allen zugute. Denn die Erforschung Seltener Erkrankungen kann auch die „Medizin von morgen“ revolutionieren.

Spenden
Wie Sie helfen können
Menschen, die an unzureichend erforschten Krankheiten leiden, brauchen schnelle Hilfe. Wir handeln. Unterstützen Sie uns mit Ihrer Spende bei unserer Forschungsoffensive für Seltene Erkrankungen!

MEHR ERFAHREN„Ohne die Förderung der Alliance4Rare wäre mein Forschungsvorhaben im universitären Alltag nicht umzusetzen.“

MEHR ERFAHREN„Ich wünsche mir mehr Aufmerksamkeit für die Erforschung und Behandlung seltener Krankheiten, mit dem Ziel, den Betroffenen eine Chance für ein besseres Leben zu ermöglichen.“

MEHR ERFAHREN„Gerade hatte ich eine Stelle an der größten Kinderklinik der Schweiz begonnen. Für die Möglichkeit exzellente Forschung und Patientenversorgung in Einklang zu bringen, bin ich zurück nach Berlin gekommen.“

MEHR ERFAHREN„Herzenswünsche e.V. fördert die Alliance4Rare, weil Hoffnung auf besser wirksame Therapien für viele Familien mit schwerkranken Kindern der wohl allergrößte Herzenswunsch ist.“

MEHR ERFAHREN„Jeder hat eine Chance auf ein normales und glückliches Leben verdient. Durch die Förderung der Alliance4Rare können Forscher wie ich hoffen, Patienten mit Seltenen Erkrankungen eine bessere Zukunft zu ermöglichen.“

MEHR ERFAHREN„Durch gezielte Nachwuchsförderung machen wir uns für bestens ausgebildete Kinderärzt:innen stark – sie sind die Zukunft der Medizin und Hoffnung für viele Familien in Berlin und weltweit.“

MEHR ERFAHREN„Die Alliance4Rare bietet mir die einmalige Chance, neben meiner fachärztlichen Tätigkeit Patienten mit Seltenen Erkrankungen ausriechend Zeit zu widmen, um die Diagnosestellung zu beschleunigen.“

MEHR ERFAHREN„Man kann sich dem Schicksal der Waisen der Medizin nicht verschließen. Schon gar nicht, wenn man sich klarmacht, wie viel hier noch durch Forschung bewegt werden kann und muss.“

Helfen Sie den Waisen der Medizin!
Die ELHKS setzt Ihre Spenden gezielt ein – damit medizinischer Fortschritt alle erreicht.
Unser kostenloser Newsletter informiert Sie über aktuelle Ausschreibungen. Veranstaltungen und Projekte zu Seltenen Erkrankungen.